|
Программные системы и вычислительные методы
Правильная ссылка на статью:
Ромашко Д.А., Медведев А.Ю.
Применение word2vec в задаче кластеризации оперонов
// Программные системы и вычислительные методы.
2018. № 1.
С. 1-6.
DOI: 10.7256/2454-0714.2018.1.25297 URL: https://nbpublish.com/library_read_article.php?id=25297
Применение word2vec в задаче кластеризации оперонов
Ромашко Дмитрий Александрович
аспирант, Дальневосточный федеральный университет
690091, Россия, Приморский край, г. Владивосток, о. Русский, поселок Аякс - 10, кампус ДВФУ, Корпус D
Romashko Dmitrii Aleksandrovich
Graduate Student, Department of Mathematical Methods in Economics, Far Eastern Federal University
690091, Russia, Primorskii krai, g. Vladivostok, o. Russkii, poselok Ayaks - 10, kampus DVFU, Korpus D

|
wintor20@gmail.com
|
|
 |
|
Медведев Александр Юрьевич
аспирант, Дальневосточный федеральный университет
690091, Россия, Приморский край, г. Владивосток, о. Русский, поселок Аякс - 10, кампус ДВФУ, Корпус D
Medvedev Aleksandr Yur'evich
Graduate Student, Department of Mathematical Methods in Economics, Far Eastern Federal University
690091, Russia, Primorskii krai, g. Vladivostok, o. Russkii, poselok Ayaks - 10, kampus DVFU, Korpus D
|
|
rf_alexmedvedev@mail.ru
|
|
|
|
DOI: 10.7256/2454-0714.2018.1.25297
Дата направления статьи в редакцию:
27-01-2018
Дата публикации:
11-02-2018
Аннотация:
В данной работе решается задача кластеризации оперонов (особых единиц генетической информации) и описывается ее использование для выделения групп оперонов со схожими функциями. Рассматривается специфика открытых баз оперонов, используемых в качестве источников исходных данных для исследования. Описываются выбор и подготовка данных для кластеризации, особенности процесса кластеризации и его связь с подходами, традиционно используемыми для анализа естественных языков. На основании проведенной кластеризации анализируется качество и состав полученных групп. Для преобразования исходных данных в вектора используется классическая реализация алгоритма word2vec и ряд особенностей исходных данных. Полученное представление кластеризуется алгоритмом DBScan на основании косинусной дистанции. Новизна предлагаемого метода связана с использованием не стандартных для исходных данных алгоритмов. Использованный подход эффективно проявляет себя при работе с большим количеством данных, не требует дополнительной разметки данных и самостоятельно формирует факторы для кластеризации. Полученные результаты показывают возможность использования предложенного подхода для реализации сервисов, позволяющих проводить сравнительный анализ бактериальных геномов.
Ключевые слова:
алгоритмы, методы, машинное обучение, word2vec, векторные представления слов, DBScan, кластеризация, опероны, анализ естественного языка, открытые базы данных
Abstract: In this article the task of clustering operons (special units of genetic information) is solved. The authors describe its use for the identification of groups of operons with similar functions. The specifics of the open bases of operons used as sources of initial data for the study are considered. The authors describe the selection and preparation of data for clustering, the features of the clustering process, and its relationship with the approaches traditionally used for the analysis of natural languages. Based on the clustering performed, the quality and composition of the obtained groups is analyzed. To convert the raw data into vectors, the classical implementation of the word2vec algorithm and a number of features of the original data are used. The resulting representation is clustered by the DBScan algorithm based on the cosine distance. The novelty of the proposed method is associated with the use of non-standard algorithms for the initial data. The approach used effectively manifests itself when working with a large amount of data, does not require additional data markup and independently forms factors for clustering. The obtained results show the possibility of using the proposed approach for the implementation of services that allow comparative analysis of bacterial genomes.
Keywords: algorithms, methods, machine learning, word2vec, word embeddings, DBScan, clustering, operons, natural language processing, open access databases
Введение
Геномные данные являются предметом многочисленных математических исследований последних лет. Эта тенденция связана с совершенствованием и удешевлением лабораторного оборудования, и, следовательно, растущей доступностью процесса секвенирования – получения данных о геноме. Благодаря этому исследования ведутся не только в пробирках, но и при помощи компьютера. Процесс начинается с экспериментальных данных, но программная обработка стала важным инструментом получения научных результатов.
Относительно простыми для обработки являются данные бактериальных геномов. Первые успехи по получению этих данных были получены в 1995 г., когда было произведено первое полногеномное секвенирование бактерии, однако в последующие годы процесс быстро набрал обороты и количество секвенированных полных бактериальных геномов показало экспоненциальный рост, на начало 2018 г., в базе данных NCBI более семи тысяч полных геномов [5]. Это привело к ситуации, когда количество доступных данных растет быстрее, чем возможности по их обработке, что в свою очередь вызывает потребность в эффективных вычислительных методах работы с данными.
Основной целью обработки геномных данных является получение информации об особенностях исследуемого организма. В геноме выделяются особые структурные единицы – гены: структурные, отвечающие за синтез белков, и функциональные, контролирующие структурные. У безъядерных организмов, например, бактерий, функционально близкие гены организуются в особые структуры – опероны, отвечающие за совместную транскрипцию входящих в них генов. Нахождение в геноме оперонов с известной функцией позволяет делать выводы о самом исследуемом организме [1].
В настоящей работе рассматривается решение задачи кластеризации оперонов при помощи существующих баз данных и методов машинного обучения. Решение этой задачи позволит эффективно выделять группы оперонов с одинаковыми биологическими функциями для последующего использования в качестве механизма определения принадлежности группы генов к тому или иному виду известных оперонов.
Анализ современных средств
Специфика работы с геномными данными подталкивает к координации лабораторий. Полученные данные используются не только в собственных исследованиях, но и впоследствии обычно выкладываются в общий доступ. Основные программные средства часто служат двойной цели – хранение геномных данных в собственных базах данных и их анализ. Программные продукты для личного использования не рассматриваются в рамках данного исследования так как предполагают наличие значительного объема предобработанных данных, что делает использование сервисов с собственной базой данных предпочтительнее при решении задач классификации оперонов.
База данных DOOR (the Database of prOkaryotic OpeRons) содержит 2072 аннотированных бактериальных генома, а также их экспериментально и вычислительно подтвержденные опероны. DOOR предоставляет доступ к базе данных и механизм поиска оперонов в предоставляемом пользователем геноме [2]. Поиск оперонов осуществляется на основе 5 основных групп факторов [1]. В основном полагаясь на межгенную дистанцию, что делает метод универсальным в ущерб точности.
Сервис OperonDB содержит 1059 аннотированных геномов бактерий и архей, а также вычислительно подтвержденные опероны. Поиск оперонов осуществляется на основе консервативных генных пар [6].
Сервис ProOpDB (the Prokaryotic Operon DataBase) содержит 1200 аннотированных бактериальных генома с вычислительно подтвержденными оперонами [11]. Поиск оперонов осуществляется при помощи MLP (multilayer perceptron neural network) алгоритм, который использует межгенную дистанцию и функциональные связи [12].
Все три сервиса используют геномные данные для поиска в них оперонов, однако не определяют их тип и принадлежность к какой-либо группе, предоставляя это пользователю. Ни один из этих сервисов не может выделить группу оперонов с одинаковой биологической функцией, что крайне неудобно если исследователь хочет получить, например, все образцы оперонов отвечающих за расщепление лактозы.
Методы исследования
Для кластеризации оперонов, необходимо представить их в виде некоторых числовых векторов. Гены – носители некоторой биологической функции, а опероны, являясь совокупностью функционально связанных генов, осуществляют некоторую совместную функцию. Если представить опероны в виде текстов, состоящих из слов – генов, тогда задача кластеризации оперонов по биологической функции становится эквивалентна задаче кластеризации текстов по смыслу.
Задача преобразования слова в вектор таким образом, чтобы слова, стоящие в схожих контекстах, имели схожие вектора эффективно решается алгоритмом word2vec [7]. Для каждого оперона необходимо максимизировать следующую функцию:
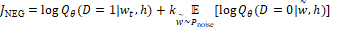
где 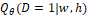 – это вероятность того, что ген – это вероятность того, что ген 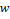 находится в опероне находится в опероне 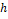 в наборе данных в наборе данных  . Эта вероятность зависит от векторов . Эта вероятность зависит от векторов 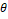 , которые и являются конечным результатом работы word2vec. Для каждого гена будет построен свой вектор . На практике , которые и являются конечным результатом работы word2vec. Для каждого гена будет построен свой вектор . На практике 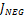 оценивается с подстановкой в оперон , оценивается с подстановкой в оперон , 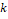 случайных слов случайных слов 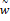 . Этот метод называется Negative Sampling. Функция . Этот метод называется Negative Sampling. Функция 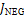 близка к максимуму тогда, когда вероятность слова близка к максимуму тогда, когда вероятность слова 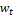 высокая, а вероятность случайных слов высокая, а вероятность случайных слов 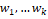 низкая [4]. низкая [4].
Вектора имеют сразу несколько ценных свойств, необходимых для решения основной задачи:
1. Гены, которые находятся в похожих оперонах, будут иметь похожие вектора.
2. Гены из одного оперона также будут иметь похожие вектора, поскольку контексты, по которым они рассчитываются, отличаются только на один ген.
3. Если вектора 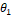 и и 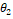 похожи, то похожи, то 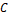 – косинусная дистанция между ними будет близка к нулю. – косинусная дистанция между ними будет близка к нулю.
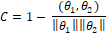
В результате работы алгоритма каждый ген будет сопоставлен -мерному вектору 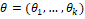 , а каждый оперон сопоставлен своему набору генов – двумерной матрице из столбцов и , а каждый оперон сопоставлен своему набору генов – двумерной матрице из столбцов и 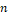 строк, где – количество входящих в оперон генов, а каждая строка – вектор соответственного гена. строк, где – количество входящих в оперон генов, а каждая строка – вектор соответственного гена.
Кластеризация оперонов на данном этапе затруднена, так как они представляют собой матрицы не равной размерности. Достаточно сжать эти матрицы в соответственный агрегированный вектор 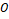 , в данной работе использовалось усреднение по каждой координате . Это возможно благодаря свойству 2), описанному выше. В итоге свойства генных векторов сохраняются и для оперонов , что позволит нам эффективно их кластеризовать, используя в качестве метрики косинусную дистанцию. , в данной работе использовалось усреднение по каждой координате . Это возможно благодаря свойству 2), описанному выше. В итоге свойства генных векторов сохраняются и для оперонов , что позволит нам эффективно их кластеризовать, используя в качестве метрики косинусную дистанцию.
Для решения задачи кластеризации необходим алгоритм, который масштабируется на большое число точек и самостоятельно определяет число кластеров. В данной работе был выбран алгоритм DBSCAN [3]. Алгоритм объединяет точки, лежащие близко друг к другу и имеющие много соседей и помечает точки с малым числом соседей как выбросы. Вычислительная сложность алгоритма составляет 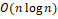 , где – число кластеризуемых точек. Преимущества DBSCAN: возможность находить кластеры любой формы, малое число параметров, устойчивость к выбросам. , где – число кластеризуемых точек. Преимущества DBSCAN: возможность находить кластеры любой формы, малое число параметров, устойчивость к выбросам.
Данные были обработаны при помощи языка программирования Python и библиотек numpy, pandas, sklearn и gensim [8, 9, 10, 13].
Результаты исследования и их обсуждение
В качестве источника исходных данных были выбраны все доступные геномы E. Coli из сервиса DOOR. Данная бактерия хорошо исследована и поэтому представляет собой репрезентативный пример. В качестве изначальной выборки взяты известные гены E. Coli, входящие в опероны. Всего 279665 генов, образующих 157528 оперонов.
Этап очистки данных крайне важен, так как все не именованные гены (hypothetical protein) описываются в исходных данных одним и тем же символом, но при этом могут быть никак между собой не связаны, что противоречит приведенной выше аналогии между геном в опероне и словом в тексте. Их исключение значительно улучшает качество модели. Опероны, состоящие из одинаковых генов, всегда группируются в один кластер, это следует из модели, так как они описываются одним и тем же вектором. Самые многочисленные среди них – состоящие из одного гена, при этом такие опероны не слишком интересны с точки зрения своей функции, их исключение облегчает читаемость полученных результатов. Таким образом в процессе очистки данных из выборки были исключены не именованные гены и все содержащие их опероны, также были исключены опероны, состоящие из одного гена. Итоговая выборка составила 86563 гена, образующих 27983 оперона.
При помощи алгоритма word2vec каждый ген был сопоставлен вектору из 50 компонент 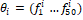 . После чего выражены вектора оперонов такой же размерности вида: . После чего выражены вектора оперонов такой же размерности вида: 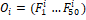 , где , где 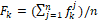 – среднее значение по всем входящим в оперон генам. – среднее значение по всем входящим в оперон генам.
После получения векторов, соответствующих оперонам, необходимо подобрать параметры для алгоритма кластеризации DBScan. Этими параметрами являются минимальное количество векторов для формирования кластера – 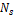 и максимальная дистанция, на которой вектора могут считаться соседними – и максимальная дистанция, на которой вектора могут считаться соседними – 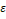 . .
Авторами DBScan рекомендовано выбирать равным удвоенному количеству компонент вектора, однако опероны редко дублируются в рамках одного генома, а всего в выборке было использовано 33 генома и 23 плазмиды, поэтому большинство кластеров будут предположительно иметь не более 30 членов, а более редкие опероны могут встречаться только в некоторых плазмидах. Поэтому было принято решение перебрать от 20 до 5 с шагом 5.
Ожидаемы средний размер кластера примерно равен 30, тогда мы ожидаем около 933 кластеров. Значит для отдельного оперона 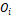 только 1 из любых 933 является соседом. На Рисунке 1 показано распределение косинусных дистанций между векторами оперонов, следует выбрать так, чтобы площадь графика слева от него была в 933 раза меньше, чем справа, был принят равным 0,3. только 1 из любых 933 является соседом. На Рисунке 1 показано распределение косинусных дистанций между векторами оперонов, следует выбрать так, чтобы площадь графика слева от него была в 933 раза меньше, чем справа, был принят равным 0,3.
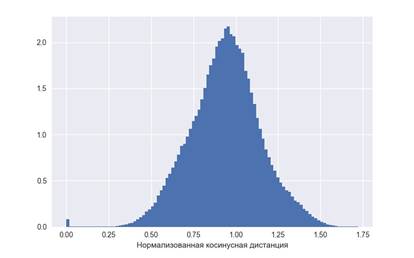
Рисунок 1 – Распределение дистанций между векторами оперонов (нормализованная гистограмма)
В результате работы DBScan с параметрами 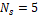 , , 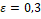 было получено 912 кластеров со средним размером 26,3 члена на кластер. 1765 оперонов алгоритм не смог определить ни в один из кластеров. При проверке результатов один из кластеров содержащий 1982 оперона был очевидно собран из неверно кластеризованных оперонов. Таким образом были удачно кластеризованы 86,6% оперонов. Легко были выделены кластеры содержащие классические опероны: лактозный оперон – кластер из 43 оперонов, триптофановый оперон – кластер из 40 оперонов. Алгоритм устойчив к изменению порядка генов в опероне и к незначительным изменениям в его структуре. было получено 912 кластеров со средним размером 26,3 члена на кластер. 1765 оперонов алгоритм не смог определить ни в один из кластеров. При проверке результатов один из кластеров содержащий 1982 оперона был очевидно собран из неверно кластеризованных оперонов. Таким образом были удачно кластеризованы 86,6% оперонов. Легко были выделены кластеры содержащие классические опероны: лактозный оперон – кластер из 43 оперонов, триптофановый оперон – кластер из 40 оперонов. Алгоритм устойчив к изменению порядка генов в опероне и к незначительным изменениям в его структуре.
Заключение
Использованные в данной работе данные были заранее значительно обработаны другими сервисами. Геномы были собраны, в них найдены гены (аннотированы) и даже размечены рамки оперонов. Все эти задачи уже достаточно хорошо решены и находятся за рамками рассмотрения данной статьи. Автоматизация процесса разделения оперонов по их функциональным особенностям, является естественным следующим этапом и должна значительно облегчить процесс анализа бактериальных геномов.
Проведенное исследование позволяет утверждать, что word2vec позволяет построить хорошее представление данных, достаточное для эффективной кластеризации оперонов. Использование алгоритмов, разработанных изначально для анализа текстов естественного языка, решает сразу ряд важных проблем анализа геномных данных: огромное количество накопленных данных становится преимуществом, не требуется самостоятельно определять факторы для обучения модели и обосновывать их значимость, не требуется предварительно размечать данные для обучения модели. Полученные результаты показывают возможность использования предложенного подхода для реализации сервисов, позволяющих проводить сравнительный функциональный анализ бактериальных геномов.
Дальнейшие исследования будут направлены на включение в анализ неаннотированных генов, дополнение входных данных другими бактериальными геномами, улучшение механизма подбора параметров модели и повышение ее точности. Также планируется создание открытой базы данных кластеров оперонов.
Библиография
1. Brouwer R.W., Kuipers O.P., Hijum S.A. The relative value of operon predictions // Brief Bioinform.-2008.-C. 367–375.
2. Cao H., Ma Q., Chen X., Xu Y. DOOR: a prokaryotic operon database for genome analyses and functional inference // Brief Bioinform.-2017.-С. 8.
3. Ester M., Kriegel Hans-Peter, Sander J., Xu X., Simoudis E., Han J., Fayyad U. M., A density-based algorithm for discovering clusters in large spatial databases with noise // Proceedings of the Second International Conference on Knowledge Discovery and Data Mining.-1996.-С. 226–231.
4. Goldberg Y., Levy O., word2vec Explained: deriving Mikolov et al.'s negative-sampling word-embedding method // arXiv.org URL: https://arxiv.org/abs/1402.3722 (дата обращения 27.01.2018)
5. Loman N.J., Pallen M.J. Twenty years of bacterial genome sequencing // Nat Rev Microbiol.-2015.-С. 787–794.
6. Mihaela Pertea, Kunmi Ayanbule, Megan Smedinghoff and Steven L. Salzberg., Prediction of Operons in Microbial Genomes // Nucleic Acids Research.-2008.-С. 479–482.
7. Mikolov T., Sutskever I., Chen K., Corrado G., Dean J., Distributed representations of words and phrases and their compositionality // Advances in neural information processing systems.-2013.-№26.-С. 3111–3119.
8. Pedregosa F., Scikit-learn: Machine Learning in Python // JMLR.-2011.-C. 2825–2830.
9. Rehurek R., Sojka P., Software Framework for Topic Modelling with Large Corpora // In proceedings of the lrec 2010 workshop on new challenges for nlp frameworks.-2010.-С. 7.
10. Stéfan van der Walt, S. Chris Colbert and Gaël Varoquaux, The NumPy Array: A Structure for Efficient Numerical Computation // Computing in Science & Engineering.-2011.-C. 22-30.
11. Taboada B., Ciria R., Martinez-Guerrer C.E., Merino E., ProOpDB: Prokaryotic Operon DataBase // Nucleic Acids Research.-2012.-№40.-С. 627-631.
12. Taboada B., Verde C., Merino E., High accuracy operon prediction method based on STRING database scores // Nucleic Acids Research.-2010.-№38.-С. 130.
13. Wes McKinney, Data Structures for Statistical Computing in Python // Proceedings of the 9th Python in Science Conference.-2010.-C. 51-56.
References
1. Brouwer R.W., Kuipers O.P., Hijum S.A. The relative value of operon predictions // Brief Bioinform.-2008.-C. 367–375.
2. Cao H., Ma Q., Chen X., Xu Y. DOOR: a prokaryotic operon database for genome analyses and functional inference // Brief Bioinform.-2017.-S. 8.
3. Ester M., Kriegel Hans-Peter, Sander J., Xu X., Simoudis E., Han J., Fayyad U. M., A density-based algorithm for discovering clusters in large spatial databases with noise // Proceedings of the Second International Conference on Knowledge Discovery and Data Mining.-1996.-S. 226–231.
4. Goldberg Y., Levy O., word2vec Explained: deriving Mikolov et al.'s negative-sampling word-embedding method // arXiv.org URL: https://arxiv.org/abs/1402.3722 (data obrashcheniya 27.01.2018)
5. Loman N.J., Pallen M.J. Twenty years of bacterial genome sequencing // Nat Rev Microbiol.-2015.-S. 787–794.
6. Mihaela Pertea, Kunmi Ayanbule, Megan Smedinghoff and Steven L. Salzberg., Prediction of Operons in Microbial Genomes // Nucleic Acids Research.-2008.-S. 479–482.
7. Mikolov T., Sutskever I., Chen K., Corrado G., Dean J., Distributed representations of words and phrases and their compositionality // Advances in neural information processing systems.-2013.-№26.-S. 3111–3119.
8. Pedregosa F., Scikit-learn: Machine Learning in Python // JMLR.-2011.-C. 2825–2830.
9. Rehurek R., Sojka P., Software Framework for Topic Modelling with Large Corpora // In proceedings of the lrec 2010 workshop on new challenges for nlp frameworks.-2010.-S. 7.
10. Stéfan van der Walt, S. Chris Colbert and Gaël Varoquaux, The NumPy Array: A Structure for Efficient Numerical Computation // Computing in Science & Engineering.-2011.-C. 22-30.
11. Taboada B., Ciria R., Martinez-Guerrer C.E., Merino E., ProOpDB: Prokaryotic Operon DataBase // Nucleic Acids Research.-2012.-№40.-S. 627-631.
12. Taboada B., Verde C., Merino E., High accuracy operon prediction method based on STRING database scores // Nucleic Acids Research.-2010.-№38.-S. 130.
13. Wes McKinney, Data Structures for Statistical Computing in Python // Proceedings of the 9th Python in Science Conference.-2010.-C. 51-56.
|
 Рус
Рус









